|
Research Interests - Gene Therapy...
Gene therapy is broadly defined as the genetic modification of a patients cell's in order to achieve a therapeutic endpoint.
Clinically, the approaches employed to reach this endpoint can be catagorised into 3 subcatagories:
- Local administration of vector - in this example the vectors is directly administored into the disease site.
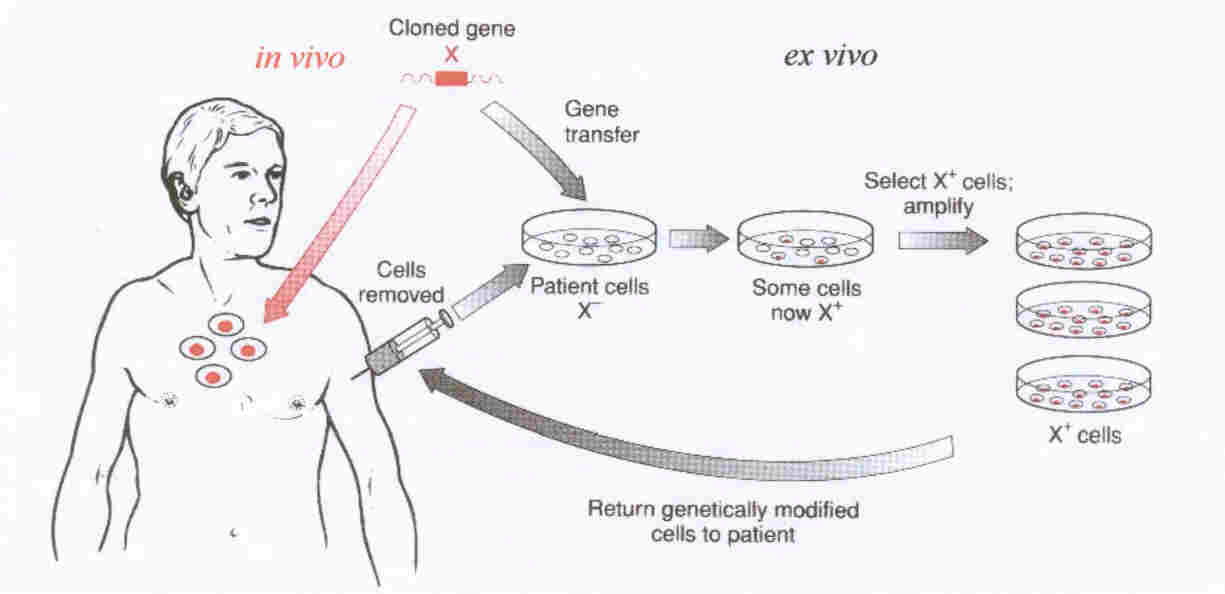
- Ex vivo gene therapy, where cells are removed from the body, genetically modified, selected and expanded outside
the body prior to reinfusion of genetically modified vector.
- In vivo gene therapy, where the vector is administored systemically into a patient in a form where it can target
to the diseased cells, mediating its therapeutic effect.
Whilst the development of ex vivo gene delivery has shown great promise in recent years, such as the recent successful
trails concerning ADA SCID, where patients showed significant clinical improvement following the ex vivo correction
of the gene encoding the gamma chain of the cytokine receptor for interleukin 2 using a retroviral vector, the results have
been somewhat overshadowed by the observation that several of the patients have gone on to develop leukemias related to insertional
mutageniesis mediated by the vector. This technology is also likely to be limited to diseased cell types that can readily
withstand being removed from the body, undergo culture, can be readily transduced and expanded, and then reinfused.
Hence it is likely to be limited to diseases of the bloodstream.
Local administration of vector would appear to be the simplest form of gene therapy for cellular diseases that do not withstand
culturing and expansion outside the human body - as it bypasses the complicated requirements of targeting to specific organs
or vascular addresses. It is not surprising, therefore, that this represents the most frequently employed delivery method
at present in the clinical setting. Where in vivo gene therapy appears to be only option, local delivery of
vector is likely to show higher levels of gene delivery to the target tissue than the systemic approach and is also likely
to allow delivery of higher dose, given that systemic delivery of vector is likely to lower the dose limiting toxicity.
Systemic in vivo gene delivery adds a further level of complexity but, hypothetically at least, represent the
most alluring option for gene delivery, especially for disseminated disease (such as in cancer). However, infusion
of vector into the bloodstream requires suppression of any immune response to the vector, abolute stability in the extracellular
compartment, and the ability to target the gene from the bloodstream to, and only to, the diseased target cells of the body.
To date this remains the holy grail of the gene therapist. For an overview of these approaches see the diagram below...
| Gene Therapy - the in vivo and ex vivo approaches |
|
|
| In vivo (red arrows) and ex vivo (black arrows) approaches to gene therapy. |
Barriers to Gene Delivery:
(i) Extracellular Barriers:
The components of the bloodstream represent
the first challenge to any gene therapy vector administered intravenously. Non-viral
complexes are generally formed in water or weak buffer solutions (e.g. 10 mM HEPES) and are held together by the electrostatic
interaction between the negatively charged DNA and positively charged lipid or polycation.
Recent studies have shown that physiological concentrations of salt (150 mM) are sufficient to cause significant dissociation
and precipitation of simple non-viral complexes such as lipoplexes and polyplexes. In
addition to increased salt concentrations, DNA vectors are introduced into an environment containing high concentrations of
serum proteins of which the major component is the 69 kDa protein albumin. A
major role of albumin is in binding and inactivating foreign or damaged materials and target them to the liver for degradation Albumin binding also caused a reversal of surface charge from positive to negative
z-potential, blocking in vitro transfection of 293 cells. Other serum proteins in lower abundance also play pivotal roles, for example, various polyplexes and lipoplexes
activate serum complement. Similarly, acute phase proteins and polyanions such
as soluble heperans can also result in complex dissociation. Other blood serum
constituents include serum nucleases and proteases, therefore any administered vector ought to be resistant to physiological
levels of such enzymes to prevent significant degradation of the therapeutic transgene before arrival within the target cell. The bloodstream also contains circulating antibodies.
Whilst specific antibody-mediated clearance is unlikely to effect non-viral systems since they are designed to be non-immunogenic,
there are indications that non-specific antibody-binding might assist clearance.
Recent work has evaluated the benefit
of surface modification of polyplexes and adenovirus using hydrophilic polymers that bearing multiple reactive groups, to
stabilise the surface of the vector by introducing a level of lateral stabilisation.
Polyplexes stabilised in this way with multivalent polymers based on poly[N-(2-hydroxypropyl)methacrylamide] (pHPMA)
show substantially increased stability to biological challenge, manifest in substantial improvements in systemic circulation
time. It is already possible to introduce targeting agents onto the surface of
these pHPMA-coated polyplexes, leading to enhanced recognition of receptor-positive cells and this is expected to enable targeting
to peripheral target sites following intravenous injection in vivo.
To read more about pHPMA and its applications, click this link.
An alternative strategy to stabilise
cationic particles to physiological environments such as in the bloodstream, is via direct chemical modification of preformed
particles, and the subsequent crosslinking (or caging) of primary amines on the polyplex surface resulting in vectors that
showed substantial stability to salt induced aggregation, nuclease degradation and aggregation in serum. Incorporation of a trigger mechanism (to release the DNA in a transcriptionally active form following its
arrival at the target cell) is to incorporate disulphide containing crosslinking reagents to exploit the intracellular reducing
environment. Such a reducing environment ought to be sufficient to reduce the
disulphide based cross linker leading to vector disassembly and release of DNA.
Non specific interactions:
Most simple non-viral vectors are positively
charged, and following intravenous infusion are likely to interact electrostatically and non-specifically with any negatively
charged cell membrane they are exposed to (red blood cells, leukocytes and endothelial cells).
Non-specific interactions with membranes results in vector depletion and even lysis.
Therefore the progress of gene delivery vectors for systemic administration will ultimately require the development
of charge neutral vectors whose non-specific membrane interactions are ablated.
Another major obstacle for systemically
applied gene delivery vectors is avoiding clearance by the reticulo-endothelial system (RES).
Circulating macrophages and liver Kupffer cells represent a principle routes of clearance of vector. The clearance can be primed by the binding of serum immunoglobulins or albumin, or can result from direct
phagocytosis, since macrophages possess scavenger receptors such as the particle receptors and polyanion receptors.
Physical barriers to extravasation
Following arrival at the target organ,
the vector must extravasate across the different vascular layers they encounter. The
first physical barrier restricting extravasation is a layer of endothelial cells, the structure of which is organ dependent. The tightest endothelial barrier is the blood-brain barrier, where the inter-endothelial
cell junctions are only a few nanometers in diameter, and there is no evidence for non-specific fluid diacytosis. Skin and muscle also contain relatively tight endothelial layers, with slightly larger inter-endothelial
cell junctions in the post-capillary venules. Fenestrated endothelium, such as
in the kidney, is much more permeable, reflecting its purpose in filtration, whilst the sinusoidal nature of the endothelium
in the liver, spleen and bone marrow permits relatively free diffusion of materials smaller than 100 nm. The tumour vasculature is often particularly leaky to blood borne macromolecules, and shows a variety of
structures with areas of hyperpermeable endothelium, possible reflecting the activities of tumour-secreted permeability factors.
Beneath the endothelial cell layer lies
the collagen basement membrane, which is continuous in most organs, and a layer of smooth muscle cells. Both of these layers will act as significant barriers to extravasation of macromolecules into the target
tissues. Finally, the target tissue will contain a significant proportion of
cells which are not the intended target cells for gene therapy, such as fibroblasts.
Transduction of such cells depletes still further the pool of vector available for transducing the target cell population.
(ii) Intracellular Barriers
Assuming a proportion of the administered
vector reaches the target cells, a therapeutic effect will only be observed assuming the vector can successfully deliver its
DNA payload to the transcriptionally active areas of the nucleus. However, the
arrival of plasmid DNA in the nucleus by passive processes is a rare event. Eukaryotic
cells rely on membrane comparmentalisation of macromolecules for many processes, and intercompartmental transfer of plasmid
DNA, in a controlled manner, represents an enormous challenge. An effective gene
delivery system would be required to bind to the target cell surface, undergo internalisation by endocytosis, escaping the
degradative pathway and allow delivery of the expression plasmid to the nucleus, where the vector must ultimately disassemble
to release the DNA in a transcriptionally active form (see diagram below).
| Intracellular barriers to transgene delivery |
|
|
Release
from Acidified Endosomes:
Many vectors enter cells through an acidified
endosome, so one of the first major barriers to successful transgene expression is the requirement to gain exit from the endosome. Although it is not absolutely certain that entry into the cytoplasm is an obligatory
step en route to the nucleus, most workers in the field assume it is so and means to enhance cytoplasmic entry are widely
pursued. Lipoplexes are thought to achieve this by entering the lipid bilayer
and by exchanging some cationic lipids with neutral ones, leading to release of the nucleic acid into the cytoplasm. Polyplexes, on the other hand, are generally not intrinsically fusogenic and must
employ some kind of molecular engineering to achieve this effect. The pH-dependent
endosomolytic action of pEI and certain types of pH-responsive dendrimers was outlined previously. Another agent that is widely used in vitro to enhance transgene
expression using polyplexes is chloroquine, although its mechanism of action is not fully delineated. Chloroquine is a lysosomotropic weak base that leads to increased osmolarity of the endosome and may induce
lysis, however it also has nuclease inhibition activity, which may be important in maintaining integrity of the DNA in both
the endosome and the cytoplasm. In addition, chloroquine binds to DNA under acid
conditions and may lead to release of some of the cationic polymers, perhaps increasing their interaction with the endosome
membrane as the pH falls. Although its precise action is unclear, chloroquine
is widely thought to promote entry of the DNA into the cytoplasm.
Another means to enhance endosome-to-cytoplasm
transfer is the use of membrane active peptides, such as those based on the influenza virus haemagglutinin protein. These N-terminal peptides are pH dependent and adopt amphipathic configurations at
low pH that are thought to insert into the endosome membrane and lead to permeabilisation.
One major advantage of these peptides over chloroquine is that they can be used, at least in principle, in vivo, since the endosomolytic agent can be covalently linked to the nucleic acid delivery vector and targeted
to specific cells. Another fusogenic peptide which has recently received attention
is melittin, a peptide derived from the venom of the honey bee. The membrane
activity of melittin appears to be independent of pH, unlike the peptides derived from the influenza haemagluttinin protein. The chemical incorporation of melittin into PEI via covalent linkage has been shown
to dramatically increase levels of gene expression using either DNA or even mRNA.
Nuclear uptake or expression in the cytoplasm?
Most present-day DNA delivery systems
can achieve appreciable levels of transgene expression only in proliferating cells.
This is a major limitation of the usefulness of such vectors, since the majority of target cells in vivo are non-cycling (even in proliferating tumours). The main
reason for this lack of efficiency in non-proliferating cells, similar to the problems faced by retrovirus, is thought to
be the barrier of the intact nuclear membrane. Whereas in proliferating cells,
the nuclear membrane disintegrates during G2-M stage of the cell division cycle, allowing vectors access to the transcriptional
machinery of the cell, this does not occur in non-proliferating cells. Entry
into the nucleus of such non-cycling, or post-mitotic cells, is restricted to passage through the nuclear pore complexes and
several research groups are presently trying to devise means to achieve this efficiently.
It is now widely accepted that delivery of DNA from the cytoplasm to the nucleus represents one of the biggest challenges
to non-viral gene delivery systems.
Mammalian cells have evolved a range
of tightly regulated mechanisms for the nuclear uptake of proteins, with the nuclear pore complex (NPC) acting as the entry
portal to the nucleus. The NPC complex is a multimeric structure comprising 50-100
proteins with an apparent molecular weight of around 125 MDa. The pore spans
the double membrane of the nucleus, and electron microscopy images suggest it contains a central transporter and so would
have an effective channel diameter around 26 nm. The pore itself may exist without
the central transporter, in which case it could have a central channel diameter around 45 nm in diameter. However, experimental data suggest that substances less than 9 nm in diameter can pass through the NPC
by diffusion, but cargoes larger than this must enter by active transport mechanisms.
The most widely used pathway for protein import appears to be mediated by basic strands of amino acids known as nuclear
localisation sequences (NLSs), which bind to cytoplasmic importin proteins, mediating translocation to the nucleus. Transport across the NPC is energy dependent and involves the activity of number of co-factors including neurotrophic factor 2 (NTF 2), the chromatin-bound guanine-nucleotide exchange
factor RCC 1 and the small GTPase Ran.
It is well established that the mechanisms
that mediate the extremely efficient import of viral DNA to the nucleus of host cells frequently harness such pathways. Therefore considerable attention has been paid to incorporating such import mechanisms
into non-viral gene delivery systems, to increase transgene expression through an improvement in delivery of DNA to the nucleus. In principle NLS can be linked to cationic polymers, or even directly to the DNA. In one of the most convincing demonstrations, Zanta et al demonstrated greatly enhanced
levels of transgene expression using linearised DNA end modified to incorporate a single NLS sequence resulting in up to a
1000 fold increase in transgene expression in rapidly dividing cells, but also a 10-30 fold enhancement of gene expression
in slowly dividing or quiescent cells. An
interesting alternative to devising means of gaining nuclear entry is to employ cytoplasmic transgene expression systems. Some authors have demonstrated the feasibility
of using plasmids encoding transgenes under control of the phage T7 polymerase, which is able to mediate cytoplasmic transcription
in mammalian cells. Encoding the T7 polymerase itself as an autogene shows good
ability to achieve cytoplasmic expression that may be effective in post-mitotic cells.
More intriguing, however, is the use of mRNA as the exogenously-applied genetic material. Historically mRNA has been regarded as being too labile for application in gene therapy strategies, although
recently it has emerged as a powerful means to gain substantial transgene expression in post-mitotic cells. Cytoplasmic expression systems offer a promising and safe alternative to simple plasmid DNA, without the
need for nuclear entry. Although these approaches, however, are effective in
non-proliferating cells, the selectivity of expression that can be conferred by using carefully chosen promoters or other
transcriptional regulatory elements is essentially lost.
Vector unpackaging
It has been suggested that further
barriers downstream of endosomal escape and into the cytoplasm and localisation within the nucleus may also be important. For example, only a small fraction of B16F10 mouse melanoma cells expressed a GFP
transgene delivered by a cationic lipid, even when tens of thousands of intact plasmids were apparent intracellularly after
24 hours of incubation, despite the cells containing more than 100 plasmids within the nucleus. This suggests a further possible
barrier of plasmid unpackaging, i.e. for gene expression to occur, the cargo DNA must dissociate from its protective vehicle. Complex dissociation can occur in the endosome network, allowing for movement from
the endosome to the nucleus. However, should nuclear targeting of the vector/DNA
complex occur, then complex dissociation must occur within the nucleus. In fact,
nuclear co-localisation of fluorescently labelled DNA and polycation has already been demonstrated. Therefore nuclear delivery of vector/DNA complexes may well be insufficient to guarantee transgene expression
unless dissociation occurs. For polyplexes, it is considered that polycation
size could influence complex dissociation. For example, one study showed that
nuclear co-localisation of DNA and polycation occurred when complexes were formed with high molecular weight polycations,
whist naked DNA alone was found in the nucleus of cells when complexes were formed with relatively short polycations. Interestingly,
maximal gene expression was observed with complexes formed using intermediate length polycations. This perhaps represents an optimal balance between protection from degradation (conferred by larger polycations)
and the ability to unpackage the DNA in a transcriptionally active formed (conferred by smaller polycations).
|